 PDF(121 KB)
PDF(121 KB)

Progress on cleaner production of vinyl chloride monomers over non-mercury catalysts
Jinli ZHANG, Nan LIU, Wei LI, Bin DAI
PDF(121 KB)
PDF(121 KB)
Progress on cleaner production of vinyl chloride monomers over non-mercury catalysts
Polyvinyl chloride (PVC) has become the third most used plastic after polyethylene and polypropylene and the worldwide demand continues to increase. Polyvinyl chloride is produced by polymerization of the vinyl chloride monomer (VCM), which is manufactured industrially via the dehydrochlorination of dichloroethane or the hydrochlorination of acetylene. Currently PVC production through the acetylene hydrochlorination method accounts for about 70% of the total PVC production capacity in China. However, the industrial production of VCM utilizes a mercuric chloride catalyst to promote the reaction of acetylene and hydrogen chloride. During the hydrochlorination, the highly toxic mercuric chloride tends to sublime, resulting in the deactivation of the catalyst and also in severe environmental pollution problems. Hence, for China, it is necessary to explore environmental friendly non-mercury catalysts for acetylene hydrochlorination as well as high efficiency novel reactors, with the aim of sustainable PVC production via the acetylene-based method. This paper presents a review of non-mercury heterogeneous and homogeneous catalysts as well as reactor designs, and recommends future work for developing cleaner processes to produce VCM over non-mercury catalysts with high activity and long stability.
polyvinyl chloride / vinyl chloride monomer / acetylene hydrochlorination / non-mercury catalysts / green chemical process
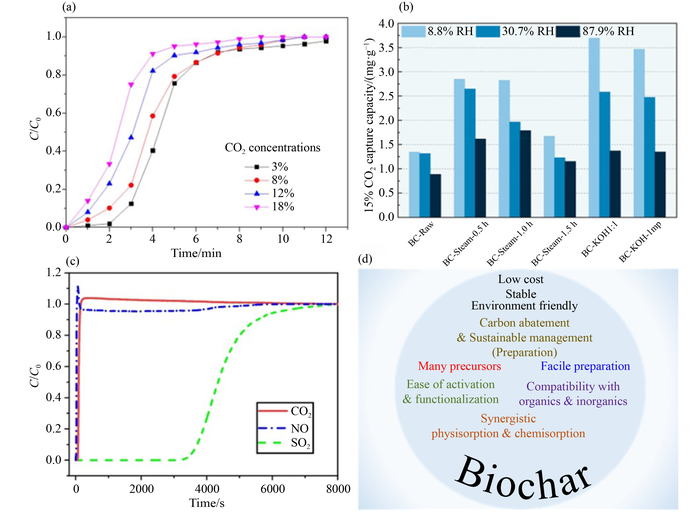
Fig.2 (a) CO2 breakthrough curves of SCK-800-1 at various CO2 inlet concentrations [12]. Reprinted with permission from ref. [12], copyright 2020, Elsevier. (b) 15% CO2 adsorption ability of wood-derived biochar (BC) samples at different relative humidity (RH) levels [3]. Reprinted with permission from ref. [3], copyright 2022, Elsevier. (BC-Raw: wood-derived raw biochar; BC-Steam: steam activated biochar; BC-KOH1:1: KOH activated biochar; BC-KOH-lmp: biochar actirated with KOH by impregnation) (c) Multicomponent breakthrough curves of CO2, NO, and SO2 through LBB20 (a kind of biochar) at 25 °C [4]. Reprinted with permission from ref. [4], copyright 2024, Springer Nature. (d) Unique advantages of biochar materials for CO2 adsorption. |
| [1] |
Ebner Martin. Ceresana research releases new comprehensive PVC market study. Newswire Today, http://www.newswiretoday.com/news/42864/, 2008-<month>11</month>-<day>18</day>
|
| [2] |
Jiang W W, Huo Y P, Yang Q, Luo Q, Li J J, Luo Y, Sun Y G. Research progress in mercury-free catalysts for hydrochiorination of acetylene. Polyvinyl Chloride, 2009, 37: 1-4 (in Chinese)
|
| [3] |
Wei F, Luo G H, Wei X B, Li X G, Qian W Z, Jin Y. CN Patent, 101670293A, 2010-<month>03</month>-<day>17</day>
|
| [4] |
Bing J L, Huang Z M. Polyvinyl Chloride (PVC) Process Technology. Beijing: Chemical Industry Press, 2008, 59(in Chinese)
|
| [5] |
Mitchenko S A, Khomutov E V, Shubin A A, Shul’ga Y M. Catalytic hydrochlorination of acetylene by gaseous HCl on the surface of mechanically pre-activated K2PtCl6 salt. Journal of Molecular Catalysis A Chemical, 2004, 212(1-2): 345-352
|
| [6] |
Hutchings G J. Gold catalysis in chemical processing. Catalysis Today, 2002, 72(1-2): 11-17
|
| [7] |
Lai C W, Jiang W W, Luo Q, Yang Q, Li J J. Study of the catalytic hydrochlorination of acetylene with nonmercuric catalytic systems. Sichuan Chemical Industry, 2007, 10: 8-11 (in Chinese)
|
| [8] |
Hutchings G. Vapor phase hydrochlorination of acetylene: correlation of catalyticn activity of supported metal chloride catalysts. Journal of Catalysis, 1985, 96(1): 292-295
|
| [9] |
Nkosi B, Coville N J, Hutchings G J. Reactivation of a supported gold catalyst for acetylene hydrochlorination. Journal of the Chemical Society. Chemical Communications, 1988, (1): 71-72
|
| [10] |
Nkosi B, Coville N J, Hutchings G J. Vapour phase hydrochlorination of acetylene with group VIII and IB metal chloride catalysts. Applied Catalysis, 1988, 43(1): 33-39
|
| [11] |
Nkosi B, Conville N J, Hutchings G J. Hydrochlorination of actylene using gold catalysts: a study of catalyst deactivation. Journal of Catalysis, 1991, 128(2): 366-377
|
| [12] |
Hutchings G J, Haruta M. A golden age of catalysis: a perspective. Applied Catalysis, A, 2005, 291: 2-5
|
| [13] |
Nkosi B, Adams M D, Coville N J, Hutchings G J. Hydrochlorination of acetylene using carbon-supported gold catalysts: a study of catalyst reactivation. Journal of Catalysis, 1991, 128(2): 378-386
|
| [14] |
Conte M, Carley A F, Hutchings G J. Reactivation of a carbon-supported gold catalyst for the hydrochlorination of acetylene. Catalysis Letters, 2008, 124(3-4): 165-167
|
| [15] |
Conte M, Carley A F, Heirene C, Willock D J, Johnston P, Herzing A A, Kiely C J, Hutchings G J. Hydrochlorination of acetylene using a supported gold catalyst: a study of the reaction mechanism. Journal of Catalysis, 2007, 250(2): 231-239
|
| [16] |
Mitchenko S A, Ananikov V P, Beletskaya I P. Mechanoactivation of acetylene hydrochlorination in the presence of K2PtCl6. Zhurnal Organicheskoi Khimii, 1998, 34: 1859-1860
|
| [17] |
Mitchenko S A. Acetylene hydrochlorination by gaseous hydrogen chloride on the surface of mechanically activated K2PtCl6 salt. Kinetics and Catalysis, 1998, 39: 859-862
|
| [18] |
Mitchenko S A, Krasnyakova T V, Mitchenko R S, Korduban A N. Acetylene catalytic hydrochlorination over powder catalyst prepared by pre-milling of K2PtCl4 salt. Journal of Molecular Catalysis A: Chemical, 2007, 275(1-2): 101-108
|
| [19] |
Strebelle M, Devos A. USPatent, 5254777, 1993-<month>10</month>-<day>19</day>
|
| [20] |
Song Q L, Wang S J, Shen B X, Zhao J G. Palladium-based catalysts for the hydrochlorination of acetylene: reasons for deactivation and its regeneration. Petroleum Science and Technology, 2010, 28(18): 1825-1833
|
| [21] |
Wang S J, Shen B X, Song Q L. Kinetics of acetylene hydrochlorination over bimetallic Au-Cu/C catalyst. Catalysis Letters, 2010, 134(1-2): 102-109
|
| [22] |
Panova S A, Shestakov K G, Temkin N O. Supported liquid-phase rhodium catalyst for acetylene hydrochlorination. Journal of the Chemical Society, Chemical Communications, 1994, (8): 977-977
|
| [23] |
Okuda N, Ueha Y, Okura K, Hisagai Y. JP Patent, 5213610A, 1993-<month>08</month>-<day>24</day>
|
| [24] |
Deng G C, Wu B X, Li T S. The reaearch on solid-liqiud catalyst using for the preparation of vinyl chloride from acetylene method. Polyvinyl Chloride, 1994, 6: 5-9 (in Chinese)
|
| [25] |
Conte M, Carley A F, Attard G, Herzing A A, Kiely C J, Hutchings G J. Hydrochlorination of acetylene using supported bimetallic Au-based catalysts. Journal of Catalysis, 2008, 257(1): 190-198
|
| [26] |
Wang S J, Shen B X, Song Q L. Kinetics of acetylene hydrochlorination over bimetallic Au-Cu/C catalyst. Catalysis Letters, 2010, 134(1-2): 102-109
|
| [27] |
Jiang W W, Yang Q, Luo Q, Li J J. CN Patent, 101249451A, 2008-<month>08</month>-<day>27</day>(in Chinese)
|
| [28] |
Yu Z Y. CN Patent, 101716528A, 2010-<month>06</month>-<day>02</day>(in Chinese)
|
| [29] |
Smith D M, Walsh P M, Slager T L. Studies of silica-supported metal chloride catalysts for the vapor-phase hydrochlorination of acetylene. Journal of Catalysis, 1968, 11(2): 113-130
|
| [30] |
Conte M, Davies T, Carley A F, Herzing A A, Kiely C J, Hutchings G J. Selective formation of chloroethane by the hydrochlorination of ethene using zinc catalysts. Journal of Catalysis, 2007, 252(1): 23-29
|
| [31] |
Enache D I, Edwards J K, Landon P, Solsona-Espriu B, Carley A F, Herzing A A, Watanabe M, Kiely C J, Knight D W, Hutchings G J. Solvent-free oxidation of primary alcohols to aldehydes using Au-Pd/TiO2. Science, 2006, 311(5759): 362-365
|
| [32] |
Hayashi T, Tanaka K, Haruta M. Selective vapor-phase epoxidation of propylene over Au/TiO2 catalysts in the presence of oxygen and hydrogen. Journal of Catalysis, 1998, 178(2): 566-575
|
| [33] |
Okazaki K, Morikawa Y, Tanaka S, Tanaka K, Kohyama M. Electronic structures of Au on TiO2(110) by first-principles calculations. Physical Review B: Condensed Matter and Materials Physics, 2004, 69(23): 235404
CrossRef
Google scholar
|
| [34] |
Cunningham D A H, Vogel W, Haruta M. Negative activation energies in CO oxidation over an icosahedral Au/Mg(OH)2 catalyst. Catalysis Letters, 1999, 63(1/2): 43-47
|
| [35] |
Bailie J E, Abdullah H A, Anderson J A, Rochester C H, Richardson N V, Hodge N, Zhang J G, Burrows A, Kiely C J, Hutchings G J. Hydrogenation of but-2-enal over supported Au/ZnO catalysts. Physical Chemistry Chemical Physics, 2001, 3(18): 4113-4121
|
| [36] |
Akita T, Tanaka K, Kohyama M, Haruta M. Analytical TEM study on structural changes of Au particles on cerium oxide using a heating holder. Catalysis Today, 2007, 122(3-4): 233-238
|
| [37] |
Kellera N, Pham-Huu C, Ledoux M J, Estournes C, Ehret G.Preparation and characterization of SiC microtubes. Applied Catalysis, A, 1999, 187(2): 255-268
|
| [38] |
Julius A N. US Patent, <patent>1812542</patent>, 1931-<month>06</month>-<day>30</day>.
|
| [39] |
Granville A P. US Patent, <patent>1934324</patent>, 1933-<month>11</month>-<day>07</day>
|
| [40] |
Armin J.US Patent, <patent>3113158</patent>, 1963-<month>12</month>-<day>03</day>
|
| [41] |
Thelen G, Bartels H, Droste W.CN Patent, 1037501A, 1989-<month>11</month>-<day>29</day>
|
| [42] |
Hutchings G J. Reactions of alkynes using heterogeneous and homogeneous cationic gold catalysts. Topics in Catalysis, 2008, 48(1-4): 55-59
|
| [43] |
Hutchings G J. Catalysis by gold. Catalysis Today, 2005, 100(1-2): 55-61
|
| [44] |
Hutchings G J, Hall M S, Carley A F, Landon P, Solsona B E, Kiely C J, Herzing A, Makkee M, Moulijin J A, Overweg A, Fierro-Gonzalez J C, Guzman J, Gates B C. Role of gold cations in the oxidation of carbon monoxide catalyzed by iron oxide-supported gold. Journal of Catalysis, 2006, 242(1): 71-81
|
| [45] |
Wei F, Wei X B, Luo G H, Qian W Z, Jin Y.CN Patent, <patent>101497046A</patent>, 2009-<month>08</month>-<day>05</day>
|



 PDF(121 KB)
PDF(121 KB)
Part of a collection:
 Fig.1 Porosity in biochar and mechanism of CO2 adsorption over biochar.Fig.2 (a) CO2 breakthrough curves of SCK-800-1 at various CO2 inlet concentrations [12]. Reprinted with permission from ref. [12], copyright 2020, Elsevier. (b) 15% CO2 adsorption ability of wood-derived biochar (BC) samples at different relative humidity (RH) levels [3]. Reprinted with permission from ref. [3], copyright 2022, Elsevier. (BC-Raw: wood-derived raw biochar; BC-Steam: steam activated biochar; BC-KOH1:1: KOH activated biochar; BC-KOH-lmp: biochar actirated with KOH by impregnation) (c) Multicomponent breakthrough curves of CO2, NO, and SO2 through LBB20 (a kind of biochar) at 25 °C [4]. Reprinted with permission from ref. [4], copyright 2024, Springer Nature. (d) Unique advantages of biochar materials for CO2 adsorption.
Fig.1 Porosity in biochar and mechanism of CO2 adsorption over biochar.Fig.2 (a) CO2 breakthrough curves of SCK-800-1 at various CO2 inlet concentrations [12]. Reprinted with permission from ref. [12], copyright 2020, Elsevier. (b) 15% CO2 adsorption ability of wood-derived biochar (BC) samples at different relative humidity (RH) levels [3]. Reprinted with permission from ref. [3], copyright 2022, Elsevier. (BC-Raw: wood-derived raw biochar; BC-Steam: steam activated biochar; BC-KOH1:1: KOH activated biochar; BC-KOH-lmp: biochar actirated with KOH by impregnation) (c) Multicomponent breakthrough curves of CO2, NO, and SO2 through LBB20 (a kind of biochar) at 25 °C [4]. Reprinted with permission from ref. [4], copyright 2024, Springer Nature. (d) Unique advantages of biochar materials for CO2 adsorption./
| 〈 |
|
〉 |

 AI Summary
AI Summary
