

1 INTRODUCTION
As one of the most studied epigenetic modifications, DNA methylation plays an essential role in the transcriptional regulation. In vertebrates, DNA methylation involves the addition of a methyl group to the 5′ position of cytosine in CpG dinucleotides [1]. 70%‒80% of the CpG dinucleotides are methylated, while CpG islands which are rich in CpG contents are usually resistant to DNA methylation [1]. DNA methylation has been associated with various cellular processes, such as embryonic development and differentiation, silencing chromosomal domains and imprinting [2‒8]. Aberrant methylation and unmethylation has been closely linked to various diseases, such as cancers [9,10]. Therefore, the genome-wide methylation detection is essential for the investigation of a broad range of diseases.
Recent progress of both experimental and computational profiling approaches for DNA methylation provides an unprecedentedly comprehensive view of the methylation landscape, and makes it possible to detect the differential methylation patterns in genome scale and in a large number of sample size [11‒22]. Especially, the development of sequencing technology at single-cell levels opens the door to exciting new fields of research [23‒27].
Excellent reviews concerning about the various experimental DNA methylation detection, preprocessing of both sequencing data and array data, the methylation biomarkers in different cancers are available [11, 28‒33]. With the rapid development of DNA methylation, the current progress of methylation detection for both pooled cells and single cells, the computational estimation of DNA methylation and the differential methylation detection methods are urgently required.
In this review, we firstly described the high-throughput detection technologies, including the bisulphite sequencing method, Illumina chiparray method, long-read sequencing method and the single cell bisulfite based techniques. Then we analyzed the three directions in computational methylation estimation methods, which include the methylation prediction, methylation expansion and methylation imputation. Furthermore, the detection methods of differential methylation patterns for sequencing and array data were presented. The potential research direction in the future were analyzed in the last chapter.
2 GENOME-WIDE PROFILING TECHNIQUES FOR DNA METHYLATION
A large variety of genome wide DNA methylation detection methods have been developed in the last few years. The developmental route could be described in the Fig.1.
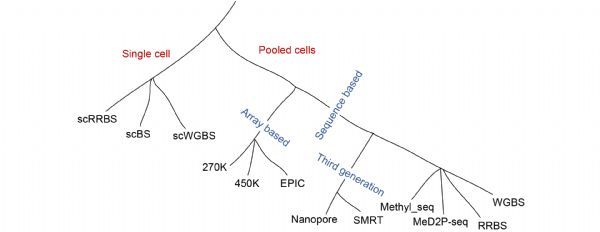
2.1 Genome-wide methylation detection for pooled cells
2.1.1 Sequencing based methods
The combination of bisulfite free or bisulfite based pretreatment approaches and sequencing based analysis method produced different detection methods, which include enzyme digestion sequencing, affinity enrichment sequencing and bisulfite sequencing methods.
Restriction enzyme-based methods cleave the unmethylated target sequences and leave the methylated DNA intact, and the following step would then reveal the locations of the unmethylated CpG sites within the recognition sites of the enzyme utilized [34]. The widely used enzyme based sequencing method is MRE-seq [35], which makes use of the differential digestion properties of isoschizomers and neoschizomers. This method has relatively low coverage of the genome and can only analyze the specific DNA sequences.
For the affinity enrichment method, it applies either methyl-CpG-binding domain proteins or antibodies to enrich methylated DNA regions. For example, MeDIP-seq utilizes an anti-methylcytosine antibody to immunoprecipitate DNA with methylated CpG sites [28], which is cost-effective and feasible when there is only a low amount of starting DNA material, but it could not discriminate methylation context.
Bisulfite sequencing approach aims to investigate specific DNA sequences in single-base resolution. The treatment of genomic DNA with sodium bisulfite results in deamination of unmethylated cytosines to uracil, and leaves the methylated cytosines unaffected [28]. Whole-genome bisulfite sequencing (WGBS) theoretically covers all the cytosine information, including low CpG density regions, such as the intergenic gene deserts and distal regulatory elements. It provides accurate binary calls of cytosine methylation status, and is considered as the golden standard method in the DNA methylation study, therefore, it has been widely used in the NIH Roadmap [36] and ENCODE projects [37]. WGBS is the most comprehensive one among the existing detection methods, while there are also two limits for the WGBS method that a considerable amount of DNA is required, and the data analysis is relatively cost and difficult. To investigate the methylome at a lower cost, the reduced representation bisulfite sequencing (RRBS) method was developed. It only sequence a fraction of the genome, which integrates MspI restriction enzyme digestion, enrichment of CPG-rich regions, bisulfite conversion and the sequencing for the analysis of specific fragments [38]. RRBS is less cost because it focuses on the enrichment of CpG-rich regions and is widely applied in methylation landscape of large-scale samples. The obvious drawback of RRBS is that it is limited to the loci containing MspI cut sites, and it exhibits a lack of coverage at intergenic and distal regulatory elements that are relatively less studied.
Because of the mappability problem of short reads, long-read sequencing method was proposed, which mainly include the nanopore sequencing from Oxford Nanopore Technologies (ONT) and single molecule real-time (SMRT) sequencing from Pacific Biosciences. PacBio’s SMRT sequencing allows the direct detection of base modification resulting from the addition of fluorescently labeled nucleotides into complementary DNA strands. It has the advantage of low requirement of input DNA, and could be applied to detect several types of epigenetic modifications, such as 6mA and 4mC [39]. In nanopore sequencing, it detects the variation in ionic current, which could distinguish the cytosine from the 5mC and 5hmC. There are mainly three steps for the nanopore sequencing, which includes base calling with canonical bases, anchoring the raw signal to a genomic reference and the identifying whether the base is modified. Its output is the probability that a base is modified at almost single-nucleotide resolution. However, because of the higher error rate and cost, these two third-generation sequencing methods are still limited for wild application.
2.1.2 Array based method
Besides the sequencing based detection methods, array based approaches are also widely used for methylation detection. For array based method, after the bisulfite conversion of genomic DNA and amplification, the converted DNA is hybridized to arrays containing predesigned probes to distinguish between methylated and unmethylated cytosines. Illumina adapts its GoldenGate BeadArray technology to interrogate DNA methylation in human genomic DNA samples, and produces three series of methylation beadchips, which includes Illumina HumanMethylation27 BeadChip (HM270K array), HumanMethylation450 BeadChip (HM450K array) and MethylationEPIC BeadChip (EPIC array). HM450K array covers more than 450,000 CpG methylation sites including 96% of the CGIs, 92% of the CGI shores and 86% of the CGI shelves [40]. The EPIC array covers more than 850,000 methylation sites which includes more than 90% of the HM450K sites plus additional CpG sites in the enhancer regions [41]. The HM450K array is the most widely used approach in the study of cancer methylome and other epigenomics, and the Cancer Genome Atlas (TCGA) consortium has mapped DNA methylation in thousands of cancer samples using array based methods [42]. The array based methods were cost-effective, but also have obvious disadvantages that the coverages of the HM270K, HM450K and EPIC array are about 0.9%, 1.5% and 2.8% of the whole genome.
2.2 Genome-wide methylation detection for single cell
The detection methods mentioned above could only provide the average measurements across bulk cell population, therefore, it is an open question that whether the methylation models based on the bulk data hold true when scrutinised on the single-cell level. To assess the methylation heterogeneity among individual cells, single-cell bisulfite based techniques are developed. The first single-cell reduced-representation bisulfite sequencing (scRRBS) was proposed by the integration of all steps to bisulfite conversion into one tube, followed by two rounds of PCR amplification and deep sequencing [43]. Because of the relatively poor coverage and the PCR-induced amplification bias about scRRBS, single-cell bisulfite sequencing (scBS-seq) was developed [23], which improved the measurement of DNA methylation at up to 48.4% of the CpG sites. In order to facilitate the full genome wide coverage and trace the allele- or strand-specific methylation differences, the technology of single-cell whole-genome bisulfite sequencing (scWGBS) was then provided to infer the epigenomic cell-state dynamics in pluripotent and differentiating cells [44].
The single cell methylation technique was firstly applied to analyze the early mammalian development, which confirmed some previously established findings and also retrieved some truly novel findings [45,46]. Besides, the application of single cell methylation studies makes significant contributions to the understanding of cancer biology and the precise cancer treatment [44,47]. But still, there is a long journey for the satisfying coverage rate for current scWGBS detection technique.
3 COMPUTATIONAL ESTIMATION OF DNA METHYLATION LEVEL
The experimental detection methods provide the most important ways to characterize the genome wide DNA methylation patterns for better understanding of the regulatory mechanisms. To improve the efficiency and accuracy of the genome wide experimental data, some computational tools that map bisulfite sequencing data and align the bisulfite-converted reads were developed [48‒50]. However, the whole genome bisulfite sequencing is expensive, labor intensive and subject to conversion bias, some immunoprecipitation sequencing methods are experimentally difficult or unfeasible in some contexts, array based methods are cost effective, but biased to gene regions and CpG islands. Therefore, the estimation of DNA methylation level with computational methods has attracted a lot attention in the last few decades. Generally, there are three directions in the computational works: prediction the methylation levels from one tissue/individual to other tissues/individuals, expanding the methylation landscape from lower coverage to higher coverage and the imputation of the single cell methylome data. The sketch map was shown in Fig.2.

{kind=link}
{kind=link}
3.1 Prediction the DNA methylation levels
Although the tissue-specific gene expression patterns that determine cell types and functions are reported to be regulated partly by tissue-specific methylation, the locus specific methylation patterns between tissues were found to be highly consistent across individuals [51,52]. Lots of works have been implemented to develop prediction models that could predict the methylation levels of different tissues [53,54].
In 2006, we developed a prediction model called MethCGI [55], which was constructed with support vector machine based on the methylation data from human brain. Applying the nucleotide sequence contents as well as transcription factor binding sites (TFBSs), MethCGI achieved specificity of 84.65% and sensitivity of 84.32% in cross validation, and it also performed quite well on some other tested tissues, such as lung, liver.
Ma et al. developed linear prediction model and SVM prediction model on peripheral blood leukocytes (PBL), atrium and artery samples based on the methylation profiling from beadarrays [56]. The methylation values either at nearby regions or somewhere on the genome that might be correlated with a particular CpG sites were used as the modeling features. Both the cross-tissue prediction validation results and independent tissue prediction results indicated that DNA methylation values were largely conserved across tissues. Their intensive research also indicated that the prediction performance would depend on the actual tissue pairs and sample size of the training data set, and the performance could be improved if the surrogate tissue would be representative of the target tissue.
Zhang et al. developed a machine learning framework for prediction and characterizing DNA methylation in mammalian genomes [57]. With the minimal set of discriminative features (OFS), such as CpG statistics, repeats, histone modifications and DNase, the methylation prediction model was trained with SVM on H1 or NPC cell. With the transfer learning strategy, the SVM model was tested on tissues different from the training tissue, and the prediction results are comparable to the corresponding results in the same cell type. Also they found that the NPC trained SVM model performed well on the MSC dataset, but performed only modestly in IMR90, which indicated that there were obvious different methylation profiling between the stem cells and the cells loss of pluripotency.
To explore the performance of cross-tissue prediction intensively, the Liang group investigated the methylation concordance between placenta and cord blood [58], whose methylation correlation is lower than other tissue pairs. Using the single-CpG-based SVM model and the multiple-CpG-based model, the locus-specific DNA methylation levels in placenta were predicted using DNA methylation levels in cord blood. The results showed that a subset of CpG sites were highly correlated between measured and predicted placenta methylation levels. Therefore, it provided a reference data to predict the methylation levels on placenta which was rarely feasible to collect in large epidemiological studies.
Our group proposed a deep learning model based on convolutional neural networks for predicting DNA methylation at single-CpG-site precision [59]. In contrast to other traditional prediction tools with predefined features, our MRCNN model trained the prediction model on H1 ESC using the raw sequence as input, and applied the model to predict the genome wide methylation levels on the normal brain white matter, lung and colon tissues. The MRCNN model trained on H1 ESC data obtained high accuracy on the prediction of the methylation levels of these tissues, which indicated that MRCNN model was robust in prediction methylation levels of different types of tissues.
3.2 Expanding the DNA methylation profiling
Some methylation detection methods only provide partial methylation landscape, it is curious to know whether it is possible to expand the detected landscape to a larger landscape, i.e., expand the methylation coverage based on the detected methylation profiling.
To better use the current thousands of 450K array data, our group developed the first expanding method for 450K array data in 2016 [60]. By integrating the sequence features in flanking regions and methylation values in 450K array data, several machine learning models were developed to predict the methylation levels of CpGs uncovered by 450K array. The proposed model expanded the coverage of Illumina 450K (~11 folds) and showed superior prediction accuracies. Then by integrating information derived from the similarity of local methylation pattern between tissues, the methylation information of flanking CpG sites and the methylation tendency of flanking DNA sequences, we updated the expanding model and applied it to 450K data of rheumatoid arthritis (RA), osteoarthritis and normal FLS [61]. Our model successfully expanded the coverage of CpG sites 18.5-fold and accounts for about 30% of all the CpGs in the human genome, and identified genes and pathways tightly related to RA pathogenesis. To improve the efficiency of expansion method, our group then presented an improved model called EAGLING with a more than 10 times faster speed compared to our previous methods [42]. In EAGLING, two features related to the methylation levels were used to build a logistic regression model, and the expanded methylation profiling, gene expression, and somatic mutation data were integrated to identify the potential biomarkers of 13 cancers in TCGA. The integrative analysis using the expanded methylation data is powerful in identifying critical genes/pathways that may serve as new therapeutic targets.
The number of interrogated CpGs in repetitive elements (RE) remains quite limited for current arrays, and the profiled CpGs in RE are generally sparse. Based on the methylation levels, the genetic information and RE information in specific flanking regions, Zheng et al. reported a random forest-based algorithm that can accurately predict genome-wide locus-specific RE methylation based on beadarray profiling [62]. The expanding model achieved satisfying performance in the validation, and achieved 2.7–3.7 times as many Alu and about 20% more LINE-1 compared with the original HM450/EPIC. Based on the expanded RE CpG loci, it enables more comprehensive differential methylation analyses and improves power to discriminate tumor from normal tissues.
With the intensive application of EPIC array, it is also an interesting field to expand methylation levels from HM450K array to EPIC array. Applying the methylation levels of 450K array as the explanatory variable, Li et al. presented an ensemble method, CUE, to get the CpG loci of EPIC that not covered in HM450K array [63]. In cross-validation, the CUE ensemble model had higher prediction accuracy and lower predicted RMSE compared with individual methods, such as KNN, logistic regression and random forest, and could obtain larger number of CpG probes passed the quality control. Accurately expanded methylation values could subsequently improve the power in downstream analysis and be helpful as new methylation arrays continue to be developed.
Considering that these expanding models concentrated on improving the overall prediction accuracy for the CpG loci while neglecting whether each locus is precisely predicted, our group developed a method for constructing precise prediction models for each single CpG locus [64]. For any interested CpG locus that presented in EPIC array but not covered in 450K array, a logistic regression algorithm called PretiMeth was built based on only one DNA methylation feature that shared the most similar methylation pattern with the CpG locus to be predicted. PretiMeth outperformed other algorithms in the prediction accuracy, and kept robust across platforms and cell types, which indicated that the precise prediction models could be probably used for reference in the probe set design when the DNA methylation beadchips update.
Besides the expanding for methylation array data, Yu et al. developed a mixture regression model (MRM) to expand the RRBS data [65]. By integrating information of neighboring CpGs and the similarities in local methylation patterns across subjects and across multiple genomic regions, they achieved satisfying accuracy on both simulated data and real RRBS data, and recovered the methylation levels of 300K CpGs in the promoter regions of chromosome 17.
The details of the main expanding models are shown in Tab.1.
Tab.1 The detail information of the main expanding models |
| Method | Algorithm | Features | Original coverage | Expanded profiling | Tool |
|---|---|---|---|---|---|
| Fan et al. [60] | SVM | a) DNA sequenceb) Neighboring methylation values | 450K | ~11 folds of 450K | / |
| Fan et al. [61] | LR | a) DNA sequenceb) Neighboring methylation valuesc) Methylation values from similarity tissues | 450K | ~18.5 folds of 450K | http://wanglab.ucsd.edu/star/LR450K/ |
| Fan et al. [42] | LR | a) Neighboring methylation valuesb) Methylation values from similarity tissues | 450K | ~18.9 folds of 450K, more than 10 times faster speed | http://114.55.236.67:8013/Integrative_Analysis/home |
| Zheng et al. [62] | RF | a) RE CpG density and RE lengthb) Smith-Waterman (SW) scorec) Number of neighboring profiled CpGsd) Genomic region of the target CpG | 450K/850K | 2.7–3.7 times Alu and about 20% more LINE-1 | http://bioconductor.org/packages/release/bioc/html/REMP.html |
| Li et al. [63] | Ensemble model | Methylation values in 450K | 450K | 850K | / |
| Tang et al. [64] | LR | Methylation feature that shared the most similar methylation pattern with the CpG locus to be predicted | 450K | 850K | https://github.com/JxTang-bioinformatics/PretiMeth |
| Yu et al. [65] | MRM | Local methylation profile from both regions and subjects | RRBS | ~ 300 K CpGs in the promoter regions of chromosome 17 | https://github.com/yuft2003/MRM |
3.3 Imputation for single-cell data
The development of scBS-seq and scRRBS makes it possible to uncover the heterogeneity and dynamics of DNA methylation. However, due to the small amount of DNA in the cells, they often result in very sparse coverage of genome-wide CpG (20%–40% for scBS-seq and 1%–10% for scRRBS-seq). For the quantitative analysis of the single-cell methylome data, the computational imputation of the single-cell methylomes becomes an important preprocessing step for the downstream analysis.
DeepCpG was the first genome-wide imputation method for single-cell DNA methylation profiles [66]. Based on the local DNA sequence and observed neighboring methylation states, DeepCpG constructed a deep neural network model to predict the binary CpG methylation states. It provided insights into how sequence composition affects methylation variability, and enabled accurate imputation of missing methylation states, which provided great facilitation for genome-wide downstream analyses.
Then Jiang et al. proposed a novel LightCpG model to identify the DNA methylation status of CpG sites in single cells [67]. Using the sequence features, structure features, positional features, and their combinations, they constructed a novel gradient boosting decision tree (GBDT) algorithm including gradient-based one-side sampling and exclusive feature bundling to reduce the training time. A series of validation experiments demonstrated that the LightCpG model could achieve outstanding performance in the imputation of DNA methylation levels with low computational complexity.
Combined with frameworks of methylation clustering, two groups developed different imputation methods separately. Kapourani and Sanguinetti proposed a Bayesian hierarchical method called Melissa which addressed data imputation on unassayed CpG loci [68]. It effectively apply both the information of neighboring CpGs and of other cells with similar methylation patterns in order to predict CpG methylation states, and output the probability distributions that fully quantify the uncertainty of the methylation prediction. It is more feasible for downstream design and analysis compared to the point-estimates provided by traditional prediction approaches. De Souza et al. proposed a novel statistical model and framework called Epiclomal to impute the missing methylation values [69]. By using a hierarchical mixture model to borrow statistical strength across cells and neighboring loci, Epiclomal could impute the inherent missing CpG methylation values more correctly on one hand, and also outperformed previous non-probabilistic methods to find the true clusters on the other hand.
Our group also implemented an imputation method based on the CatBoost gradient boosting model to impute the single-cell methylation states [70]. Besides the DNA sequence information, intracellular neighboring methylation states, and the similarity of local methylation patterns between units, CaMelia was designed to construct a separate model for each cell which could avoid excessive smoothing of all cells and learn the unicellular nature of cells. Experimental validation results on real single-cell methylation datasets indicated that CaMelia yielded significant imputation performance improvement over previous methods. In the downstream analysis of cell-type identification, our CaMelia model could help to discover more intercellular differentially methylated loci that were masked in sparse raw data, and improve the identification of cell subpopulations.
4 THE IDENTIFICATION OF DIFFERENTIAL METHYLATION PATTERN
The identification of differential methylation pattern (DMP) is an effective way to discover the abnormal methylated genes closely related to tissue specificity and the development of diseases, such as cancer. A comprehensive series of methods identifying the DMP were developed according to the whole genome sequencing data and array data [71‒82].
4.1 DMP detection for sequencing data
For the sequencing methylation data, the Zhang group developed the first user-friendly tool named CpG_MPs for identification and analysis of the methylation patterns of genomic regions from bisulfite sequencing data [71]. CpG_MPs defined the interested regions using the methylation status of neighboring CpGs by hotspot extension algorithm, and the differentially methylated regions across paired or multiple samples are identified with a combinatorial Shannon entropy algorithm. Applying the tool on human bisulfite sequencing data during cellular differentiation, some potentially functional regions related to cellular differentiation were retrieved.
Then Park et al. presented a statistical package named methylSig to analyze the difference between disease and control samples [72]. Taking the read coverage and biological variation into consideration, methlSig applied a beta-binomial approach to identify methylation differences for CpG loci or regions across defined groups. It had high sensitivity and potential to identify the important regions in diseased samples.
The Wang group proposed a novel integrative statistical framework named M&M which combined MeDIP-seq and MRE-seq data to detect differentially methylated regions [73]. By modeling the relationship between DNA methylation level, CpG content, and expected sequencing reads in defined region, M&M constructed a statistical model to compute a probability score and identified the differential methylated regions.
As the methods above did not provide accurate statistical inference, Korthauer et al. developed an inferential approach based on a pooled null distribution [74]. Based on a nested autoregressive correlated error structure, it fit a generalized least squares regression model and constructed a region-level statistic to detect the DMP. The detection results based on both Monte Carlo simulation and experimental data indicated that it could improve the specificity and sensitivity of lists of regions and control the false discovery rate accurately.
4.2 DMP detection for array data
For array methylation data, the mean and the median methylation values between the compared groups were applied for the differential methylation detection. The Zhang group developed FastDMA which aimed to identify significantly differentially methylated probes and differentially methylated region [75]. Based on a uniformed statistical model, analysis of covariance was used to analyze the Beadchip array data for the DMP scanning. Many differentially methylated sites in different types of cancer were identified on three large-scale DNA methylation datasets from TCGA.
To better use the correlation of nearby CpG sites, Shen et al. presented a novel Bayesian framework named DMRMark for detecting DMRs from methylation array data [76]. By combining the constrained Gaussian mixture model and the biological knowledge with the nonhomogeneous hidden Markov model, DMRMark could detect the DMRs without the requirement of predefined boundaries or decision windows, and could detect DMRs from even a single pair of samples or unpaired samples.
Methods applicable to both of the sequencing and array methylation data were also proposed. Zhang et al. developed an unsupervised approach named QDMRs to identify DMRs with Shannon entropy [77]. It is applied to MeDIP-chip data of human tissues and RRBS data of mouse, and identified some potential functional regions, which indicated that QDMR was a platform-free and species-free model. Besides the regions of CpG islands and promoter regions, Lee et al. presented a wavelet-based functional mixed model to detect the DMRs in other genomic regions [78]. By accommodating spatial correlations across genomes and correlations between samples through functional random effects, they developed the framework which could be applied to different settings and had more power in the DMR detection. Then Denault et al. improved a powerful wavelet-based method called Fast Functional Wavelet detect the DMRs fast [79]. Combining the results of theoretical null distribution of Bayes factors, it achieved fast emulation of the test statistic and reduced the computational time considerably.
5 DISCUSSION AND FUTURE PERSPECTIVES
More and more studies and results indicated the great importance of DNA methylation changes across many types of solid tumors, hematological malignancies, autoimmune diseases, metabolic and neurological disorders as well as the aging [83‒94]. Many aberrant DNA methylation patterns have been demonstrated to be specific of certain diseases, and the presence of differential methylation patterns are potentially diagnostic, prognostic and predictive markers. Although a lot of enormous progress in understanding the role of DNA methylation in different diseases, only few methylated genes or functional elements serve as clinically relevant cancer biomarkers [95]. Therefore, it is urgent to identify the most effective methylation biomarkers for the specific disease diagnosis, prognosis or response to treatment [11,96]. Rapid development of global DNA methylation profiling techniques has allowed our understanding of diverse aspects of genomic DNA methylation in health and disease, the bottleneck in DNA methylation advances has shifted from data generation to data analysis [32].
As the most promising direction for DNA methylation, the current single-cell methylation measurement techniques are seriously plagued by the low coverage of CpGs assayed. Therefore, it is essential to develop versatile imputation tools and address the inherent sparsity of the single-cell methylation data for quantitative analysis of the whole genome. The current imputation models achieved satisfying overall imputation accuracies, however, the biologists are more interested in whether the methylation status of a specific CpG locus could be accurately imputed. Therefore, it would be meaningful to develop the precise imputation model and provide the confidence level for a specific CpG locus of a specific cell.
The detection of differential methylation pattern would identify many abnormal genes/elements in different diseases, but a series of validation experiments are necessary to confirm the accuracy and reproducibility. In the validation, the receiver operator characteristic (ROC) curve was used to determine the sensitivity and specificity of the candidate biomarker genes [97,98], Cox proportional hazard model or Kaplan–Meier survival analysis were applied to discover their recurrence and response to treatment [99‒101]. Furthermore, the proper integration of multi-omics is also an effective way for deeper insights into disease etiology [102,103], and machine learning methods offer novel techniques to integrate and analyze the various omics data enabling the discovery of new biomarkers [104,105]. As there are three distinct features for the multi-omics data: the large number of features relative to the number of samples, the strong dependencies between features in different omics, and the intrinsic lower dimensional representation of the omics data, more versatile and powerful machine learning models need to be carefully developed in the future [106].