Background: In recent years, since the molecular docking technique can greatly improve the efficiency and reduce the research cost, it has become a key tool in computer-assisted drug design to predict the binding affinity and analyze the interactive mode.
Results: This study introduces the key principles, procedures and the widely-used applications for molecular docking. Also, it compares the commonly used docking applications and recommends which research areas are suitable for them. Lastly, it briefly reviews the latest progress in molecular docking such as the integrated method and deep learning.
Conclusion: Limited to the incomplete molecular structure and the shortcomings of the scoring function, current docking applications are not accurate enough to predict the binding affinity. However, we could improve the current molecular docking technique by integrating the big biological data into scoring function.

The impressive conversational and programming abilities of ChatGPT make it an attractive tool for facilitating the education of bioinformatics data analysis for beginners. In this study, we proposed an iterative model to fine-tune instructions for guiding a chatbot in generating code for bioinformatics data analysis tasks. We demonstrated the feasibility of the model by applying it to various bioinformatics topics. Additionally, we discussed practical considerations and limitations regarding the use of the model in chatbot-aided bioinformatics education.
Background: The coronavirus disease 2019 (COVID-19) is rapidly spreading in China and more than 30 countries over last two months. COVID-19 has multiple characteristics distinct from other infectious diseases, including high infectivity during incubation, time delay between real dynamics and daily observed number of confirmed cases, and the intervention effects of implemented quarantine and control measures.
Methods: We develop a Susceptible, Un-quanrantined infected, Quarantined infected, Confirmed infected (SUQC) model to characterize the dynamics of COVID-19 and explicitly parameterize the intervention effects of control measures, which is more suitable for analysis than other existing epidemic models.
Results: The SUQC model is applied to the daily released data of the confirmed infections to analyze the outbreak of COVID-19 in Wuhan, Hubei (excluding Wuhan), China (excluding Hubei) and four first-tier cities of China. We found that, before January 30, 2020, all these regions except Beijing had a reproductive number
Conclusions: We suggest that rigorous quarantine and control measures should be kept before early March in Beijing, Shanghai, Guangzhou and Shenzhen, and before late March in Hubei. The model can also be useful to predict the trend of epidemic and provide quantitative guide for other countries at high risk of outbreak, such as South Korea, Japan, Italy and Iran.

The principles and molecular mechanisms underlying biological pattern formation are difficult to elucidate in most cases due to the overwhelming physiologic complexity associated with the natural context. The understanding of a particular mechanism, not to speak of underlying universal principles, is difficult due to the diversity and uncertainty of the biological systems. Although current genetic and biochemical approaches have greatly advanced our understanding of pattern formation, the progress mainly relies on experimental phenotypes obtained from time-consuming studies of gain or loss of function mutants. It is prevailingly considered that synthetic biology will come to the application age, but more importantly synthetic biology can be used to understand the life. Using periodic stripe pattern formation as a paradigm, we discuss how to apply synthetic biology in understanding biological pattern formation and hereafter foster the applications like tissue engineering.

The CRISPR-Cas9 system, naturally a defense mechanism in prokaryotes, has been repurposed as an RNA-guided DNA targeting platform. It has been widely used for genome editing and transcriptome modulation, and has shown great promise in correcting mutations in human genetic diseases. Off-target effects are a critical issue for all of these applications. Here we review the current status on the target specificity of the CRISPR-Cas9 system.


Background: The extremely small amount of DNA in a cell makes it difficult to study the whole genome of single cells, so whole-genome amplification (WGA) is necessary to increase the DNA amount and enable downstream analyses. Multiple displacement amplification (MDA) is the most widely used WGA technique.
Results: Compared with amplification methods based on PCR and other methods, MDA renders high-quality DNA products and better genome coverage by using phi29 DNA polymerase. Moreover, recently developed advanced MDA technologies such as microreactor MDA, emulsion MDA, and micro-channel MDA have improved amplification uniformity. Additionally, the development of other novel methods such as TruePrime WGA allows for amplification without primers.
Conclusion: Here, we reviewed a selection of recently developed MDA methods, their advantages over other WGA methods, and improved MDA-based technologies, followed by a discussion of future perspectives. With the continuous development of MDA and the successive update of detection technologies, MDA will be applied in increasingly more fields and provide a solid foundation for scientific research.

Background: Histone modifications are major factors that define chromatin states and have functions in regulating gene expression in eukaryotic cells. Chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq) technique has been widely used for profiling the genome-wide distribution of chromatin-associating protein factors. Some histone modifications, such as H3K27me3 and H3K9me3, usually mark broad domains in the genome ranging from kilobases (kb) to megabases (Mb) long, resulting in diffuse patterns in the ChIP-seq data that are challenging for signal separation. While most existing ChIP-seq peak-calling algorithms are based on local statistical models without account of multi-scale features, a principled method to identify scale-free board domains has been lacking.
Methods: Here we present RECOGNICER (Recursive coarse-graining identification for ChIP-seq enriched regions), a computational method for identifying ChIP-seq enriched domains on a large range of scales. The algorithm is based on a coarse-graining approach, which uses recursive block transformations to determine spatial clustering of local enriched elements across multiple length scales.
Results: We apply RECOGNICER to call H3K27me3 domains from ChIP-seq data, and validate the results based on H3K27me3’s association with repressive gene expression. We show that RECOGNICER outperforms existing ChIP-seq broad domain calling tools in identifying more whole domains than separated pieces.
Conclusion: RECOGNICER can be a useful bioinformatics tool for next-generation sequencing data analysis in epigenomics research.

Background: Polygenic risk score (PRS) derived from summary statistics of genome-wide association studies (GWAS) is a useful tool to infer an individual’s genetic risk for health outcomes and has gained increasing popularity in human genetics research. PRS in its simplest form enjoys both computational efficiency and easy accessibility, yet the predictive performance of PRS remains moderate for diseases and traits.
Results: We provide an overview of recent advances in statistical methods to improve PRS’s performance by incorporating information from linkage disequilibrium, functional annotation, and pleiotropy. We also introduce model validation methods that fine-tune PRS using GWAS summary statistics.
Conclusion: In this review, we showcase methodological advances and current limitations of PRS, and discuss several emerging issues in risk prediction research.

Background: Mendelian randomization (MR) analysis has become popular in inferring and estimating the causality of an exposure on an outcome due to the success of genome wide association studies. Many statistical approaches have been developed and each of these methods require specific assumptions.
Results: In this article, we review the pros and cons of these methods. We use an example of high-density lipoprotein cholesterol on coronary artery disease to illuminate the challenges in Mendelian randomization investigation.
Conclusion: The current available MR approaches allow us to study causality among risk factors and outcomes. However, novel approaches are desirable for overcoming multiple source confounding of risk factors and an outcome in MR analysis.

In May 1985 there was at University of California Santa Cruz an influential meeting that was the first serious discussion of sequencing the entire human genome. The author was one of the participants and described the meeting and related issues.

Background: The recent development of metagenomic sequencing makes it possible to massively sequence microbial genomes including viral genomes without the need for laboratory culture. Existing reference-based and gene homology-based methods are not efficient in identifying unknown viruses or short viral sequences from metagenomic data.
Methods: Here we developed a reference-free and alignment-free machine learning method, DeepVirFinder, for identifying viral sequences in metagenomic data using deep learning.
Results: Trained based on sequences from viral RefSeq discovered before May 2015, and evaluated on those discovered after that date, DeepVirFinder outperformed the state-of-the-art method VirFinder at all contig lengths, achieving AUROC 0.93, 0.95, 0.97, and 0.98 for 300, 500, 1000, and 3000 bp sequences respectively. Enlarging the training data with additional millions of purified viral sequences from metavirome samples further improved the accuracy for identifying virus groups that are under-represented. Applying DeepVirFinder to real human gut metagenomic samples, we identified 51,138 viral sequences belonging to 175 bins in patients with colorectal carcinoma (CRC). Ten bins were found associated with the cancer status, suggesting viruses may play important roles in CRC.
Conclusions: Powered by deep learning and high throughput sequencing metagenomic data, DeepVirFinder significantly improved the accuracy of viral identification and will assist the study of viruses in the era of metagenomics.

Background: Genome-wide association studies (GWAS) have succeeded in identifying tens of thousands of genetic variants associated with complex human traits during the past decade, however, they are still hampered by limited statistical power and difficulties in biological interpretation. With the recent progress in expression quantitative trait loci (eQTL) studies, transcriptome-wide association studies (TWAS) provide a framework to test for gene-trait associations by integrating information from GWAS and eQTL studies.
Results: In this review, we will introduce the general framework of TWAS, the relevant resources, and the computational tools. Extensions of the original TWAS methods will also be discussed. Furthermore, we will briefly introduce methods that are closely related to TWAS, including MR-based methods and colocalization approaches. Connection and difference between these approaches will be discussed.
Conclusion: Finally, we will summarize strengths, limitations, and potential directions for TWAS.

Background: Herpes simplex virus type 1 (HSV-1) is a ubiquitous infectious pathogen that widely affects human health. To decipher the complicated human-HSV-1 interactions, a comprehensive protein-protein interaction (PPI) network between human and HSV-1 is highly demanded.
Methods: To complement the experimental identification of human-HSV-1 PPIs, an integrative strategy to predict proteome-wide PPIs between human and HSV-1 was developed. For each human-HSV-1 protein pair, four popular PPI inference methods, including interolog mapping, the domain-domain interaction-based method, the domain-motif interaction-based method, and the machine learning-based method, were optimally implemented to generate four interaction probability scores, which were further integrated into a final probability score.
Results: As a result, a comprehensive high-confidence PPI network between human and HSV-1 was established, covering 10,432 interactions between 4,546 human proteins and 72 HSV-1 proteins. Functional and network analyses of the HSV-1 targeting proteins in the context of human interactome can recapitulate the known knowledge regarding the HSV-1 replication cycle, supporting the overall reliability of the predicted PPI network. Considering that HSV-1 infections are implicated in encephalitis and neurodegenerative diseases, we focused on exploring the biological significance of the brain-specific human-HSV-1 PPIs. In particular, the predicted interactions between HSV-1 proteins and Alzheimer’s-disease-related proteins were intensively investigated.
Conclusion: The current work can provide testable hypotheses to assist in the mechanistic understanding of the human-HSV-1 relationship and the anti-HSV-1 pharmaceutical target discovery. To make the predicted PPI network and the datasets freely accessible to the scientific community, a user-friendly database browser was released at http://www.zzdlab.com/HintHSV/index.php.

Background: Mutational signatures computed from somatic mutations, allow an in-depth understanding of tumorigenesis and may illuminate early prevention strategies. Many studies have shown the regulation effects between somatic mutation and gene expression dysregulation.
Methods: We hypothesized that there are potential associations between mutational signature and gene expression. We capitalized upon RNA-seq data to model 49 established mutational signatures in 33 cancer types. Both accuracy and area under the curve were used as performance measures in five-fold cross-validation.
Results: A total of 475 models using unconstrained genes, and 112 models using protein-coding genes were selected for future inference purposes. An independent gene expression dataset on lung cancer smoking status was used for validation which achieved over 80% for both accuracy and area under the curve.
Conclusion: These results demonstrate that the associations between gene expression and somatic mutations can translate into the associations between gene expression and mutational signatures.

Background: Alzheimer’s disease (AD) is one of the most popular tauopathies. Neurofibrillary tangles and senile plaques are widely recognized as the pathological hallmarks of AD, which are mainly composed of tau and β-amyloid (Aβ) respectively. Recent failures of drugs targeting Aβ have led scientists to scrutinize the crucial impact of tau in neurodegenerative diseases. Mutated or abnormal phosphorylated tau protein loses affinity with microtubules and assembles into pathological accumulations. The aggregation process closely correlates to two amyloidogenic core of PHF6 (306VQIVYK311) and PHF6* (275VQIINK280) fragments. Moreover, tau accumulations display diverse morphological characteristics in different diseases, which increases the difficulty of providing a unifying neuropathological criterion for early diagnosis.
Results: This review mainly summarizes atomic-resolution structures of tau protein in the monomeric, oligomeric and fibrillar states, as well as the promising inhibitors designed to prevent tau aggregation or disaggregate tau accumulations, recently revealed by experimental and computational studies. We also systematically sort tau functions, their relationship with tau structures and the potential pathological processes of tau protein.
Conclusion: The current progress on tau structures at atomic level of detail expands our understanding of tau aggregation and related pathology. We discuss the difficulties in determining the source of neurotoxicity and screening effective inhibitors. We hope this review will inspire new clues for designing medicines against tau aggregation and shed light on AD diagnosis and therapies.

Background: Genome-wide association studies (GWAS) have identified thousands of genomic non-coding variants statistically associated with many human traits and diseases, including cancer. However, the functional interpretation of these non-coding variants remains a significant challenge in the post-GWAS era. Alternative polyadenylation (APA) plays an essential role in post-transcriptional regulation for most human genes. By employing different poly(A) sites, genes can either shorten or extend the 3′-UTRs that contain cis-regulatory elements such as miRNAs or RNA-binding protein binding sites. Therefore, APA can affect the mRNA stability, translation, and cellular localization of proteins. Population-scale studies have revealed many inherited genetic variants that potentially impact APA to further influence disease susceptibility and phenotypic diversity, but systematic computational investigations to delineate the connections are in their earliest states.
Results: Here, we discuss the evolving definitions of the genetic basis of APA and the modern genomics tools to identify, characterize, and validate the genetic influences of APA events in human populations. We also explore the emerging and surprisingly complex molecular mechanisms that regulate APA and summarize the genetic control of APA that is associated with complex human diseases and traits.
Conclusion: APA is an intermediate molecular phenotype that can translate human common non-coding variants to individual phenotypic variability and disease susceptibility.

Background: Microfluidic systems have advantages such as a high throughput, small reaction volume, and precise control of the cellular position and environment. These advantages have allowed microfluidics to be widely used in several fields of synthetic biology in recent years.
Results: In this article, we reviewed the microfluidic-based methods for synthetic biology from two aspects: the construction of synthetic gene circuits and the analysis of synthetic gene systems. We used some examples to illuminate the progresses and challenges in the steps of synthetic gene circuits construction and approaches of gene expression analysis with microfluidic systems.
Conclusion: Comparing to traditional methods, microfluidic tools promise great advantages in the synthetic genetic circuit building and analysis process. Moreover, new microfluidic systems together with the mathematical modeling of synthetic circuits or consortiums are desirable to perform complex genetic circuit construction and understand the natural gene regulation in cells and population interactions better.

Background: Genome-wide association studies (GWAS) have been widely adopted in studies of human complex traits and diseases.
Results: This review surveys areas of active research: quantifying and partitioning trait heritability, fine mapping functional variants and integrative analysis, genetic risk prediction of phenotypes, and the analysis of sequencing studies that have identified millions of rare variants. Current challenges and opportunities are highlighted.
Conclusion: GWAS have fundamentally transformed the field of human complex trait genetics. Novel statistical and computational methods have expanded the scope of GWAS and have provided valuable insights on the genetic architecture underlying complex phenotypes.

Background: The precise and efficient analysis of single-cell transcriptome data provides powerful support for studying the diversity of cell functions at the single-cell level. The most important and challenging steps are cell clustering and recognition of cell populations. While the precision of clustering and annotation are considered separately in most current studies, it is worth attempting to develop an extensive and flexible strategy to balance clustering accuracy and biological explanation comprehensively.
Methods: The cell marker-based clustering strategy (cmCluster), which is a modified Louvain clustering method, aims to search the optimal clusters through genetic algorithm (GA) and grid search based on the cell type annotation results.
Results: By applying cmCluster on a set of single-cell transcriptome data, the results showed that it was beneficial for the recognition of cell populations and explanation of biological function even on the occasion of incomplete cell type information or multiple data resources. In addition, cmCluster also produced clear boundaries and appropriate subtypes with potential marker genes. The relevant code is available in GitHub website (huangyuwei301/cmCluster).
Conclusions: We speculate that cmCluster provides researchers effective screening strategies to improve the accuracy of subsequent biological analysis, reduce artificial bias, and facilitate the comparison and analysis of multiple studies.

Background: A novel coronavirus (the SARS-CoV-2) has been identified in January 2020 as the causal pathogen for COVID-19 , a pandemic started near the end of 2019. The Angiotensin converting enzyme 2 protein (ACE2) utilized by the SARS-CoV as a receptor was found to facilitate the infection of SARS-CoV-2, initiated by the binding of the spike protein to human ACE2.
Methods: Using homology modeling and molecular dynamics (MD) simulation methods, we report here the detailed structure and dynamics of the ACE2 in complex with the receptor binding domain (RBD) of the SARS-CoV-2 spike protein.
Results: The predicted model is highly consistent with the experimentally determined structures, validating the homology modeling results. Besides the binding interface reported in the crystal structures, novel binding poses are revealed from all-atom MD simulations. The simulation data are used to identify critical residues at the complex interface and provide more details about the interactions between the SARS-CoV-2 RBD and human ACE2.
Conclusion: Simulations reveal that RBD binds to both open and closed state of ACE2. Two human ACE2 mutants and rat ACE2 are modeled to study the mutation effects on RBD binding to ACE2. The simulations show that the N-terminal helix and the K353 are very important for the tight binding of the complex, the mutants are found to alter the binding modes of the CoV2-RBD to ACE2.

Background: As one of the representative protein materials, protein nanocages (PNCs) are self-assembled supramolecular structures with multiple advantages, such as good monodispersity, biocompatibility, structural addressability, and facile production. Precise quantitative functionalization is essential to the construction of PNCs with designed purposes.
Results: With three modifiable interfaces, the interior surface, outer surface, and interfaces between building blocks, PNCs can serve as an ideal platform for precise multi-functionalization studies and applications. This review summarizes the currently available methods for precise quantitative functionalization of PNCs and highlights the significance of precise quantitative control in fabricating PNC-based materials or devices. These methods can be categorized into three groups, genetic, chemical, and combined modification.
Conclusion: This review would be constructive for those who work with biosynthetic PNCs in diverse fields.

Background: The hierarchical three-dimensional (3D) architectures of chromatin play an important role in fundamental biological processes, such as cell differentiation, cellular senescence, and transcriptional regulation. Aberrant chromatin 3D structural alterations often present in human diseases and even cancers, but their underlying mechanisms remain unclear.
Results: 3D chromatin structures (chromatin compartment A/B, topologically associated domains, and enhancer-promoter interactions) play key roles in cancer development, metastasis, and drug resistance. Bioinformatics techniques based on machine learning and deep learning have shown great potential in the study of 3D cancer genome.
Conclusion: Current advances in the study of the 3D cancer genome have expanded our understanding of the mechanisms underlying tumorigenesis and development. It will provide new insights into precise diagnosis and personalized treatment for cancers.

Background: As parts of the cis-regulatory mechanism of the human genome, interactions between distal enhancers and proximal promoters play a crucial role. Enhancers, promoters, and enhancer-promoter interactions (EPIs) can be detected using many sequencing technologies and computation models. However, a systematic review that summarizes these EPI identification methods and that can help researchers apply and optimize them is still needed.
Results: In this review, we first emphasize the role of EPIs in regulating gene expression and describe a generic framework for predicting enhancer-promoter interaction. Next, we review prediction methods for enhancers, promoters, loops, and enhancer-promoter interactions using different data features that have emerged since 2010, and we summarize the websites available for obtaining enhancers, promoters, and enhancer-promoter interaction datasets. Finally, we review the application of the methods for identifying EPIs in diseases such as cancer.
Conclusions: The advance of computer technology has allowed traditional machine learning, and deep learning methods to be used to predict enhancer, promoter, and EPIs from genetic, genomic, and epigenomic features. In the past decade, models based on deep learning, especially transfer learning, have been proposed for directly predicting enhancer-promoter interactions from DNA sequences, and these models can reduce the parameter training time required of bioinformatics researchers. We believe this review can provide detailed research frameworks for researchers who are beginning to study enhancers, promoters, and their interactions.

Background: The COVID-19 pandemic has become a formidable threat to global health and economy. The coronavirus SARS-CoV-2 that causes COVID-19 is known to spread by human-to-human transmission, and in about 40% cases, the exposed individuals are asymptomatic which makes it difficult to contain the virus.
Methods: This paper presents a modified SEIR epidemiological model and uses concepts of optimal control theory for analysis of the effects of intervention methods of the COVID19. Fundamentally the pandemic intervention problem can be viewed as a mathematical optimization problem as there are contradictory outcomes in terms of reduced infection and fatalities but with serious economic downturns.
Results: Concepts of optimal control theory have been used to determine the optimal control (intervention) levels of i) social contact mitigation and suppression, and ii) pharmaceutical intervention modalities, with minimum impacts on the economy. Numerical results show that with optimal intervention policies, there is a significant reduction in the number of infections and fatalities. The computed optimum intervention policy also provides a timeline of systematic enforcement and relaxation of stay-at-home regulations, and an estimate of the peak time and number of hospitalized critical care patients.
Conclusion: The proposed method could be used by local and state governments in planning effective strategies in combating the pandemic. The optimum intervention policy provides the necessary lead time to establish necessary field hospitals before getting overwhelmed by new patient arrivals. Our results also allow the local and state governments to relax social contact suppression guidelines in an orderly manner without triggering a second wave.

Background: Genome-wide association studies (GWASs) have identified thousands of genetic variants that are associated with many complex traits. However, their biological mechanisms remain largely unknown. Transcriptome-wide association studies (TWAS) have been recently proposed as an invaluable tool for investigating the potential gene regulatory mechanisms underlying variant-trait associations. Specifically, TWAS integrate GWAS with expression mapping studies based on a common set of variants and aim to identify genes whose GReX is associated with the phenotype. Various methods have been developed for performing TWAS and/or similar integrative analysis. Each such method has a different modeling assumption and many were initially developed to answer different biological questions. Consequently, it is not straightforward to understand their modeling property from a theoretical perspective.
Results: We present a technical review on thirteen TWAS methods. Importantly, we show that these methods can all be viewed as two-sample Mendelian randomization (MR) analysis, which has been widely applied in GWASs for examining the causal effects of exposure on outcome. Viewing different TWAS methods from an MR perspective provides us a unique angle for understanding their benefits and pitfalls. We systematically introduce the MR analysis framework, explain how features of the GWAS and expression data influence the adaptation of MR for TWAS, and re-interpret the modeling assumptions made in different TWAS methods from an MR angle. We finally describe future directions for TWAS methodology development.
Conclusions: We hope that this review would serve as a useful reference for both methodologists who develop TWAS methods and practitioners who perform TWAS analysis.

Background: A traditional Chinese medicine formula, Youdujing (YDJ) ointment, is widely used for treatment of human papilloma virus-related diseases, such as cervical cancer. However, the underlying mechanisms by which active compounds of YDJ alleviates cervical cancer are still unclear.
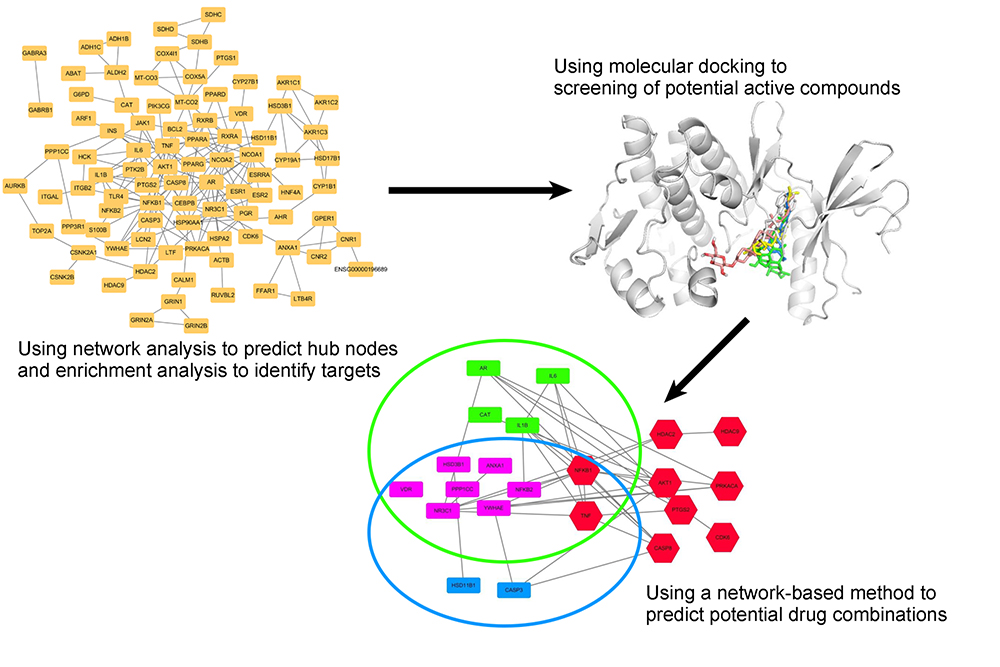
Methods: We applied a comprehensive network pharmacology approach to explore the key mechanisms of YDJ by integrating potential target identification, network analysis, and enrichment analysis into classical molecular docking procedures. First, we used network and enrichment analyses to identify potential therapeutic targets. Second, we performed molecular docking to investigate the potential active compounds of YDJ. Finally, we carried out a network-based analysis to unravel potentially effective drug combinations.
Results: Network analysis yielded four potential therapeutic targets: ESR1, NFKB1, TNF, and AKT1. Molecular docking highlighted that these proteins may interact with four potential active compounds of YDJ: E4, Y2, Y20, and Y21. Finally, we found that Y2 or Y21 can act alone or together with E4 to trigger apoptotic cascades via the mitochondrial apoptotic pathway and estrogen receptors.
Conclusion: Our study not only explained why YDJ is effective for cervical cancer treatment, but also lays a strong foundation for future clinical studies based on this traditional medicine.
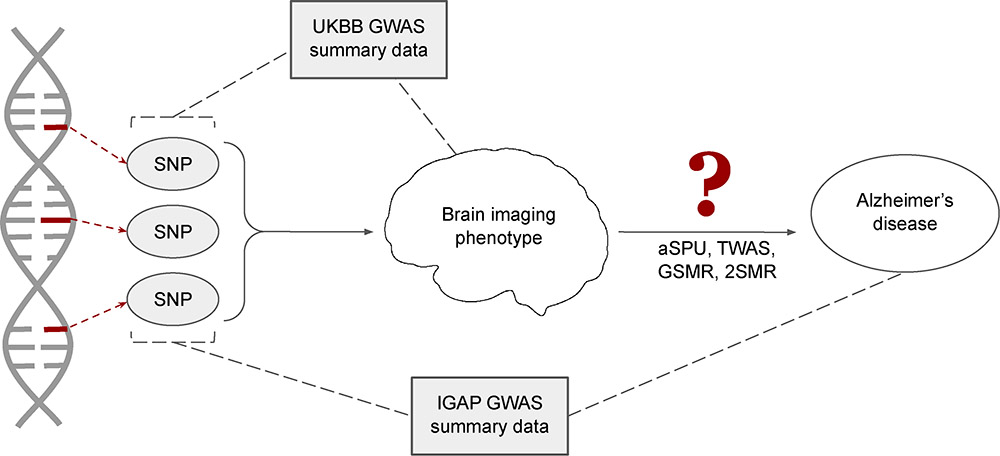
Background: Genome wide association studies (GWAS) have identified many genetic variants associated with increased risk of Alzheimer’s disease (AD). These susceptibility loci may effect AD indirectly through a combination of physiological brain changes. Many of these neuropathologic features are detectable via magnetic resonance imaging (MRI).
Methods: In this study, we examine the effects of such brain imaging derived phenotypes (IDPs) with genetic etiology on AD, using and comparing the following methods: two-sample Mendelian randomization (2SMR), generalized summary statistics based Mendelian randomization (GSMR), transcriptome wide association studies (TWAS) and the adaptive sum of powered score (aSPU) test. These methods do not require individual-level genotypic and phenotypic data but instead can rely only on an external reference panel and GWAS summary statistics.
Results: Using publicly available GWAS datasets from the International Genomics of Alzheimer’s Project (IGAP) and UK Biobank’s (UKBB) brain imaging initiatives, we identify 35 IDPs possibly associated with AD, many of which have well established or biologically plausible links to the characteristic cognitive impairments of this neurodegenerative disease.
Conclusions: Our results highlight the increased power for detecting genetic associations achieved by multiple correlated SNP-based methods, i.e., aSPU, GSMR and TWAS, over MR methods based on independent SNPs (as instrumental variables).
Availability: Example code is available at https://github.com/kathalexknuts/ADIDP.